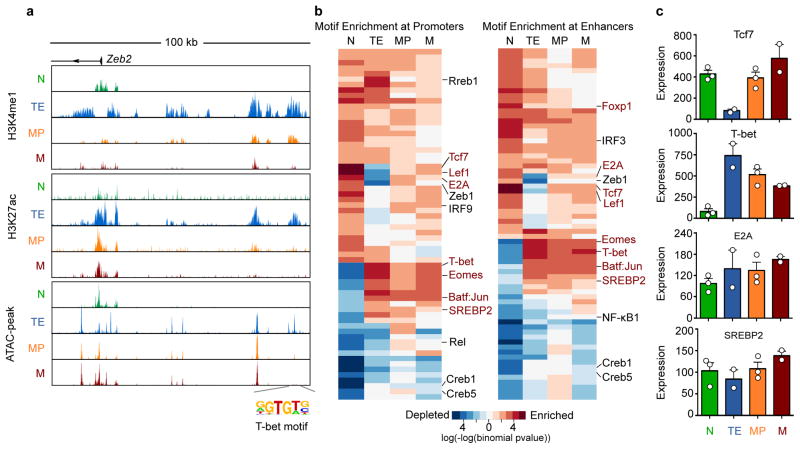
Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation
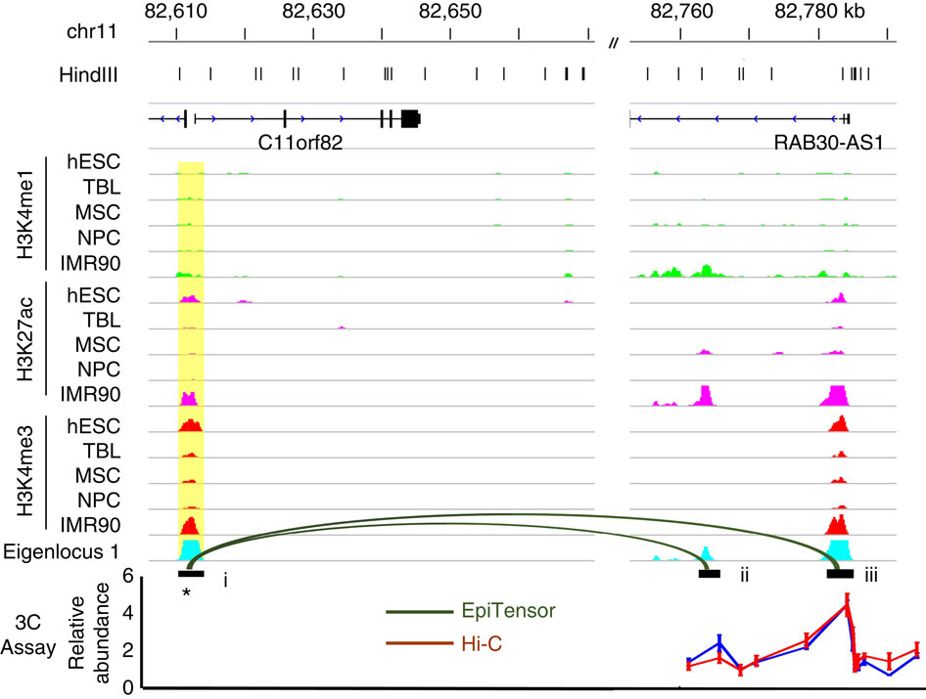
Constructing 3D interaction maps from 1D epigenomes
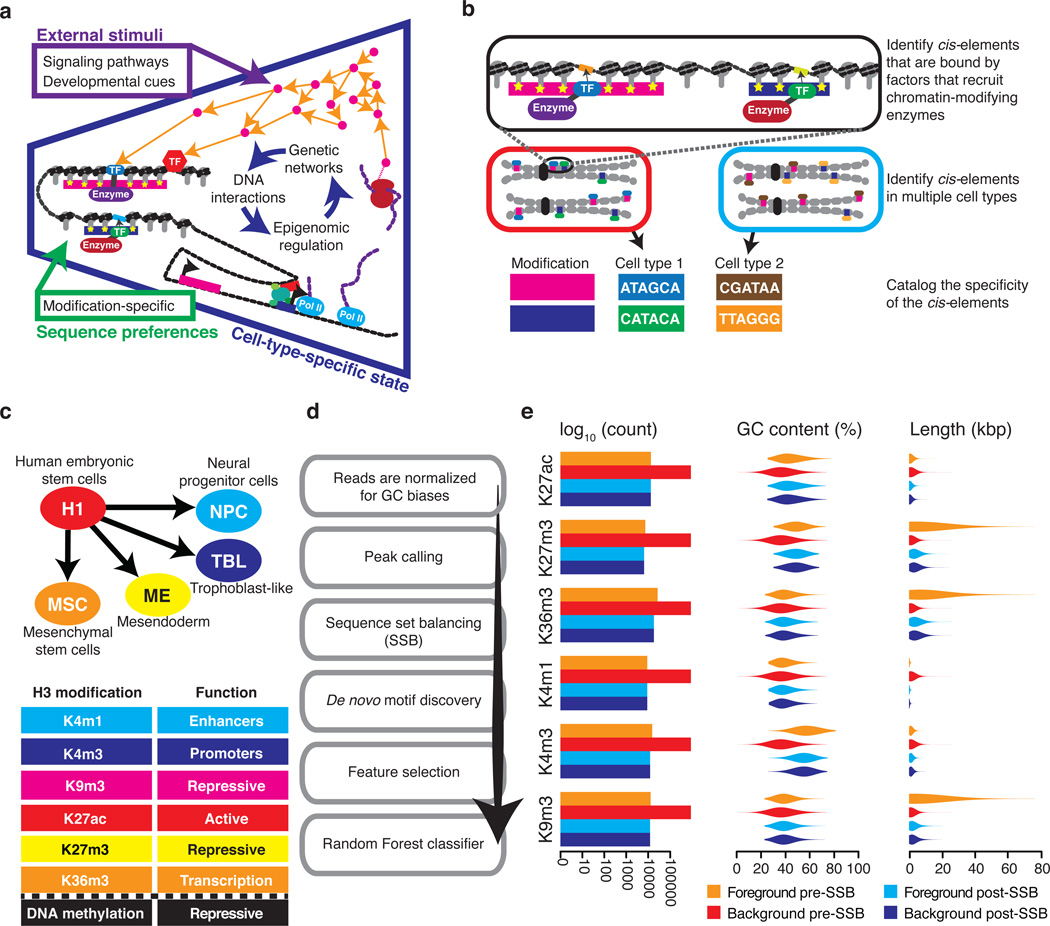
Predicting the human epigenome from DNA motifs
We are interested in uncovering the regulatory architecture of the genome and understanding the underlying biophysical principles at genomic and molecular levels. Our research is highly interdisciplinary. The computational and theoretical work are tightly coupled with experimental investigation. The focus areas in our lab include (1) Characterizing and engineering the binding specificity of protein recognition; (2) Deciphering the regulatory grammar encoded in the genome; (3) Understanding the relationship between structure and function of the 3D genome; (4) System-level study of cell state specification, cell fate decision and cell type conversion.
1. Characterizing and engineering the binding specificity of protein recognition. We are interested in understanding how the specificity of protein binding is achieved. Our recent work on characterizing the recognition of histone modifications by reader proteins reveals that the binding specificity is determined by the physiochemical patterns defined by the post-translational modifications (PTMs) on the histone residues rather than specific combinations of PTMs (Hard et al., Science Advances, 2018). A computational model characterizing the energetic patterns of the binding interface can successfully predict the reader proteins’ recognition of PTMs that are not even included in the training data. This study suggests that, unlike the genetic code represented by a simple table, “histone code” is decided by the physiochemical properties of the histone peptides and can be interpreted by a computational model. This way, combinations of any PTMs on histone tails including those newly discovered ones can be included as part of the “histone code” because reader proteins recognize the physiochemical properties of the binding interface rather than specific PTMs. We further demonstrated that the binding affinity and specificity of the reader proteins can be efficiently and effectively engineered by combining interpretable deep learning models and high throughput mutation screening (Parkinson et al, JCIM, 2020). We are currently improving the interpretable deep learning models to engineer antibodies and other proteins.
2. Deciphering the regulatory grammar encoded in the genome. The cell is a fine regulated system and the regulatory information is encoded in the genome. For example, transcription factors (TFs) recognize specific DNA motifs and the binding of TFs to particular loci determine which genes are transcribed. While epigenetic modifications including DNA methylation and histone modifications are highly locus-specific, the modifying enzymes do not have strong DNA sequence preference or do not bind to DNA at all. How the locus specificity of epigenetic modifications is achieved and maintained is a key question to answer in epigenetics. We have developed machine learning methods to identify and catalog DNA motifs associated with DNA methylation and histone modifications (Whitaker et al., Nature Methods, 2015; Wang et al., NAR, 2019; Ngo et al., PNAS, 2019). These motifs, called epi-motifs, are tightly associated with epigenetic modifications and disruption of these motifs can lead to significant change of the regional epigenetic state. We propose that the proteins or ncRNAs recognizing these epi-motifs function as pioneer factors and their binding to specific loci initiate recruitment of co-factors and epigenetic modifying enzymes We are currently working on identifying these factors using biochemical assays and confirming their functionalities. We are also developing new AI models to uncover additional regulatory rules encoded in the genome.
3. Understanding the relationship between structure and function of the 3D genome. We are interested in studying how the chromatin structure and functional activities are associated with and impact each other (Zhu et al., Nature Comm., 2016; Zhang et al., Nature Comm., 2016). For example, we developed the first unsupervised learning method to predict 3D contacts of promoter-enhancer, promoter-promoter and enhancer-enhancer from the epigenomic data (Zhu et al., Nature Comm., 2016). Recently we started to investigate the structural and functional importance of genomic regions that form many 3D contacts (called hubs) (Ding et al., Science Advances, 2021). The majority of these hub loci are epigenetically quiescent that have no or low epigenetic signals and whose importance are often overlooked. We observed that genetic variations on hub can significantly change their 3D contact numbers, particularly in disease cells. Deleting hub loci can cause cell death and significant global change of chromatin structure. Importantly, expressions of genes and promoter-enhancer contacts located distal in the linear genome from the deleted hub are often significantly altered. The collective and/or synergistic effect of these alterations cause phenotypic changes such as cell viability. Our analyses suggest that hubs play important roles in forming and maintaining the proper chromatin organization for normal cellular functions. An implication of these observations is to allow develop potent “one-drug-multiple-target” therapeutics by editing the disease-specific non-coding loci. We are currently expanding the scale of this study and working on uncovering the underlying mechanisms.
4. System-level study of cell state specification, cell fate decision and cell type conversion. We are interested in uncovering key regulators that decide cell state, cell type and conversion between cell types. We have developed various systems biology methods to model the epigenetic landscape of cell states and identify key regulators of specific cell state/type. Recently, we developed Taiji, a method that integrates DNA motif, gene expression, and epigenomic data, to construct genetic networks in individual samples (Zhang et al., Science Advances, 2019). Based on the genetic network, the global importance of each gene is assessed by Personalized PageRank algorithm weighted by gene expression and regulatory strength. Analysis of the key regulators in various tissues and different time stages reveal the transcriptional waves in tissue specification during mouse embryonic development. An intriguing observation is that there is no TF activated in specific time stage across all tissues, indicating a distributed timing system to coordinate development of different tissues (Zhang et al., Science Advances, 2019). We have also applied Taiji to study CD8+ T cell differentiation (Yu et al., Nature Immunology, 2017), other biological systems and patient samples. Furthermore, identification of cell type specific regulators can guide developing cocktails to convert one cell type to another, which is useful in generating cells needed for therapeutic treatment. We are currently improving the computational model and expanding the scope of the systems biology study.